I'm seeking a Python code that can initially construct a 3D grid surrounding my molecule and subsequently populate the vacant space within the grid with dummy atoms. I've provided an image illustrating my desired outcome, where each sphere represents an atom. I aim to replicate this using Python, with the only difference being that I want to create a cavity at the center of the cube to accommodate specific molecules. The total number of atoms required is in the hundreds. While I can generate a 3D grid, I lack a clear approach on how to position dummy atoms at each grid point. Any guidance or ideas on achieving this would be greatly appreciated.
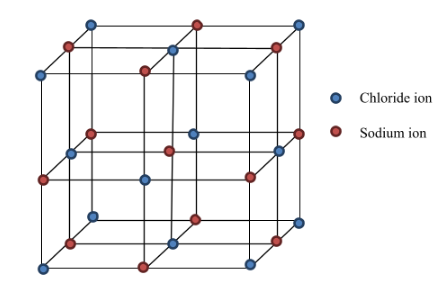
I expect the cube as shown in above image with relatively higher number of atoms, (hundreds or thousands), along with a cavity at center of the cube. Finally I also expect to get coordinates of each grid point in a separate text file.

Are you populating a pdb file or something else like a .gro?
In most cases, I believe what you'll want is to construct a unit cell. There is some helpful information here about building one: https://computecanada.github.io/molmodsim-md-theory-lesson-novice/04-Periodic_Boundary/index.html
If using gromacs, it will be able to easily constract you one using the command: gmx editconf -f coord.pdb -o outputcoord.pdb -d 1.0 -bt cubic
here you're saying "take the input coordinate file, generate an output coordinate file with a cubic box, where the edges of the box are 1.0 nm away from the edges of the molecule."
You can adjust the -d value to be 0 if you want each molecule at the edge of the box, however it's generally recommended to leave a little space for the solvent, and to avoid any periodic boundary condition issues that will arise by having your atoms sitting on the cube's points.
adding atoms into the box can be done using the gromacs insert-molecules command, which you can see more of here: https://manual.gromacs.org/current/onlinehelp/gmx-insert-molecules.html
If there is a specific reason for not wanting to use any MD software, the python script would have to comprehend your pdf file, find the outermost ions and write functions to insert the molecules into the spaces. You may also want to run a minimization of the coordinates in order to evenly distribute them according to their forcefields. You can find more on minimization runs here: https://adkgromacstutorial.readthedocs.io/en/latest/emin.html
I wouldn't write a bespoke Python script for that when gromacs can do it for you and pdb files are strictly formatted enough to be a headache to rewrite. No need to reinvent the wheel.